KCNJ10 Antibody
-
货号:CSB-PA012048GA01HU
-
规格:¥3,900
-
其他:
产品详情
-
产品描述:
The KCNJ10 polyclonal antibody is produced by immunizing rabbits with the human KCNJ10 protein. It exists as an IgG. It can cross-react with human, mouse, and rat KCNJ10 protein. Antigen affinity purification of this antibody gives this antibody relatively high purity. It is available for use in ELISA and WB assays. The target protein KCNJ10 plays a prominent role in the maintenance of resting membrane potential (RMP), extracellular K+ uptake, cell volume regulation, and facilitation of glutamate uptake, as well as in neurodegenerative diseases.
-
Uniprot No.:P78508
-
基因名:KCNJ10
-
别名:KCNJ10; ATP-sensitive inward rectifier potassium channel 10; ATP-dependent inwardly rectifying potassium channel Kir4.1; Inward rectifier K(+ channel Kir1.2; Potassium channel, inwardly rectifying subfamily J member 10
-
宿主:Rabbit
-
反应种属:Human,Mouse,Rat
-
免疫原:Human KCNJ10
-
免疫原种属:Homo sapiens (Human)
-
抗体亚型:IgG
-
纯化方式:Antigen Affinity Purified
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:PBS with 0.1% Sodium Azide, 50% Glycerol, pH 7.3. -20°C, Avoid freeze / thaw cycles.
-
产品提供形式:Liquid
-
应用范围:ELISA,WB
-
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:May be responsible for potassium buffering action of glial cells in the brain. Inward rectifier potassium channels are characterized by a greater tendency to allow potassium to flow into the cell rather than out of it. Their voltage dependence is regulated by the concentration of extracellular potassium; as external potassium is raised, the voltage range of the channel opening shifts to more positive voltages. The inward rectification is mainly due to the blockage of outward current by internal magnesium. Can be blocked by extracellular barium and cesium. In the kidney, together with KCNJ16, mediates basolateral K(+) recycling in distal tubules; this process is critical for Na(+) reabsorption at the tubules.
-
基因功能参考文献:
- results show that a glia-neuron interaction at the perisomatic space of LHb is involved in setting the neuronal firing mode in models of a major psychiatric disease. Kir4.1 in the LHb might have potential as a target for treating clinical depression. PMID: 29446379
- A novel mutation in the KCNJ10 and previously characterized mutation in KCNT1 were identified in boy with seizures and neurodevelopmental delay. KCNJ10 L218F mutation associated with disease resulted in reduced Kir current. PMID: 28747464
- rs17375748, rs1130183, rs12133079 and rs1186688 associated with sudden infant death syndrome PMID: 28520217
- kcnj10 plays a role in K(+) recycling across the basolateral membrane in corresponding nephron segments and in generating negative membrane potential--{REVIEW} PMID: 27122539
- Previous research had shown that Kir4.1 protein autoantibodies were specific for multiple sclerosis but they found that they weren't. PMID: 27074083
- This study identifies potential SNPs of KCNJ10 gene that may contribute to seizure susceptibility and anti-epileptic drug resistance. PMID: 25874548
- disruption of cav-1 decreases basolateral K(+) channel activity and depolarizes the cell membrane potential in the DCT1 at least in part by suppressing the stimulatory effect of c-Src on Kcnj10 PMID: 25848073
- anti-KIR4.1 antibody levels differed in multiple sclerosis patients during relapse and remission; as such, they may represent a marker of disease exacerbation PMID: 25392324
- This study showed that rs2486253, but not rs61822012, polymorphism of KCNJ10 gene was associated with childhood idiopathic generalized epilepsy. PMID: 25008907
- we confirmed the presence of anti-Kir4.1 antibodies in multiple sclerosis patients, but at a much lower prevalence than previously reported. PMID: 24756568
- KCNJ10 SNP is not associated with nonsyndromic enlargement of vestibular aqueduct in Chinese patients. PMID: 25372295
- No KIR4.1-specific antigen is detected in serum or cerebrospinal fluid of multiple sclerosis (MS) patients; the target antigen of MS remains elusive. PMID: 25008548
- This study observed a decrease of astroglial KIR4.1 but not glial fibrillary acidic protein IR. In chronic inactive and remyelinating MS lesions, KIR4.1 IR was restored on astrocytes and found in a subset of presumably new myelinating oligodendrocytes. PMID: 24777949
- study provides an explanation for the pathophysiology of the p.A167V KCNJ10 mutation, which had not been considered pathogenic on its own; findings provide evidence for functional cooperation of KCNJ10 and KCNJ16; in vitro ascertainment of KCNJ10 function may necessitate co-expression with KCNJ16 PMID: 24193250
- Mislocalization of the Kir4.1 channels contributes to renal salt wasting. PMID: 24561201
- KCNJ10rs1130183 did not contribute to risk of seizure susceptibility. PMID: 24378235
- Study confirms that EAST syndrome can be caused by many different mutations in KCNJ10 that significantly reduce K+ conductance. PMID: 21849804
- Ordered disorder of the astrocytic dystrophin-associated protein complex in the norm and pathology. PMID: 24014171
- Serum antibodies to KIR4.1 are found in the majority of children with acquired demylinating disease but not in children with other diseases or in healthy controls. PMID: 24415573
- the modulation of tyrosine phosphorylation of KCNJ10 should play a role in regulating membrane transport function in DCT1. PMID: 23873931
- We found no evidence for a significant association between mutations of KCNJ10 and FOXI1 with SLC26A4 in Pendred syndrome/enlarged vestibular aqueducts. PMID: 23965030
- The results of this study indicated that alterations in expression of Kir4.1 occurring in epilepsy-associated lesions are possibly influenced by the local inflammatory environment and in particular by the inflammatory cytokine IL-1beta. PMID: 23270518
- Oligodendrocyte precursor cells establish themselves progressively through postnatal upregulation of Kir4.1 potassium channels. PMID: 23392672
- The subcellular co-localisation of K(ir)4.1 and AQP4 in the supporting cells of the cochlea described in this study resembles that of the astroglia of the central nervous system and the glial Mueller cells in the retina. PMID: 22802001
- This study demonistrated that Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. PMID: 22878665
- No KCNJ10 mutations were present in bilateral deafness patients with inner ear malformation. PMID: 22412181
- Downregulation of Kir4.1 channels aggravates the visual impairment caused by the initial photoreceptor degeneration. PMID: 22055109
- Gain-of-function defects in Kir4.1 causes dysfunction in astrocytic-dependent potassium buffering and contributes to autism/epilepsy phenotype by altering neuronal excitability and synaptic function. PMID: 21458570
- extracellular volume recordings indicate that compromised K(+) spatial buffering in brain underlies the epilepsy phenotype associated with human KCNJ10 mutations PMID: 21748805
- Role of KCNJ10 function in the physiology of proximal and possibly also the distal retina. Impact of KCNJ10 mutations on the electroretinogram in four unrelated patients with EAST syndrome. PMID: 21300747
- Mutations in the K+ channel gene KCNJ10 (Kir4.1) cause the autosomal recessive EAST syndrome which is characterized by epilepsy, ataxia, sensorineural deafness, and a salt-wasting tubulopathy. PMID: 21221631
- This study suggests that the SNPs within the kcnj10 genes we examined do not play a major role in schizophrenia in the Han Chinese population. PMID: 20933057
- CaR decreases cell surface expression of Kir4.1 channels via a mechanism that involves Galpha(q) and caveolin. PMID: 21084311
- Perturbed pH gating may underlie the loss of channel function for the disease-associated mutant Kir4.1 channels and may have important physiologic consequences. [review] PMID: 21088294
- The Kir4.1 channel transgene plays a role in setting the membrane potential of glial cells and in maintaining potassium permeability in glial-conditioned Kir4.1 knock-out mice. PMID: 21106816
- SLC26A4, FOXI1 and KCNJ10 are not major determinants in unilateral deafness and enlarged vestibular aqueduct PMID: 20621367
- When expressed in CHO and HEK293 cells, the KCNJ10 mutations R65P, G77R, and R175Q caused a marked impairment of channel function PMID: 20651251
- Variations in the AQP4 and the KCNJ10/KCNJ9 region are likely to be associated with temporal lobe epilepsy. PMID: 19864112
- molecular analysis on chromosome 1q as a candidate gene for Type 2 diabetes in Pima Indians PMID: 12401729
- Arg271Cys missense variation in KCNJ10 (or a nearby variation) is related to general seizure susceptibility in humans. PMID: 15120748
- Our results support previous evidence that the common KCNJ10 Arg271Cys missense variation influences seizure susceptibility of common IGE syndromes. PMID: 15725393
- Calcium-sensing receptor interacts directly with Kir4.1 and Kir4.2 and can decrease their currents. PMID: 17122384
- The results showed that the expression of Kir 4.1 mRNA and protein, as well as the Kir 4.1 immunoreactivity score (IRS), increased markedly with increasing pathologic grade. PMID: 18191638
- identifY previously unidentified KCNJ10 missense or nonsense mutations on both alleles in all subjects affected by a unique human syndrome, and establish the essential role of basolateral K(+) channels in renal electrolyte homeostasis. PMID: 19289823
- Mutations in KCNJ10 cause a specific disorder. Our findings indicate that KCNJ10 plays a major role in renal salt handling and possibly also in blood-pressure maintenance and its regulation. PMID: 19420365
- mutations in the inwardly rectifying K(+) channel gene KCNJ10 are associated with nonsyndromic hearing loss in carriers of SLC26A4 mutations with an EVA/PS phenotype. PMID: 19426954
显示更多
收起更多
-
相关疾病:Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SESAMES)
-
亚细胞定位:Membrane; Multi-pass membrane protein. Basolateral cell membrane.
-
蛋白家族:Inward rectifier-type potassium channel (TC 1.A.2.1) family, KCNJ10 subfamily
-
组织特异性:Expressed in kidney (at protein level).
-
数据库链接:
HGNC: 6256
OMIM: 602208
KEGG: hsa:3766
STRING: 9606.ENSP00000357068
UniGene: Hs.408960
Most popular with customers
-
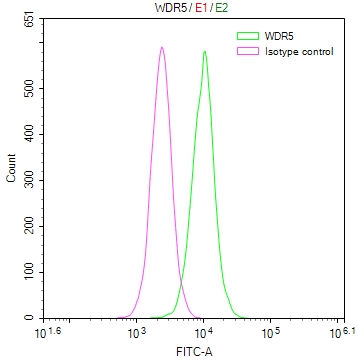
-
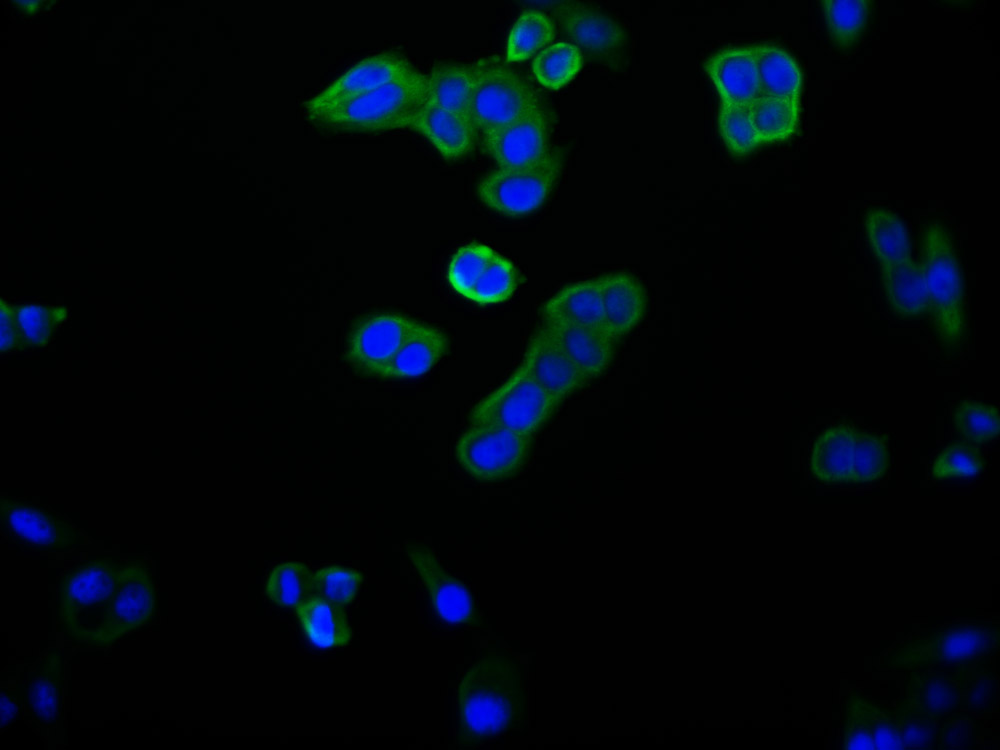
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-