KCNQ4 Antibody
-
货号:CSB-PA320879
-
规格:¥1100
-
图片:
-
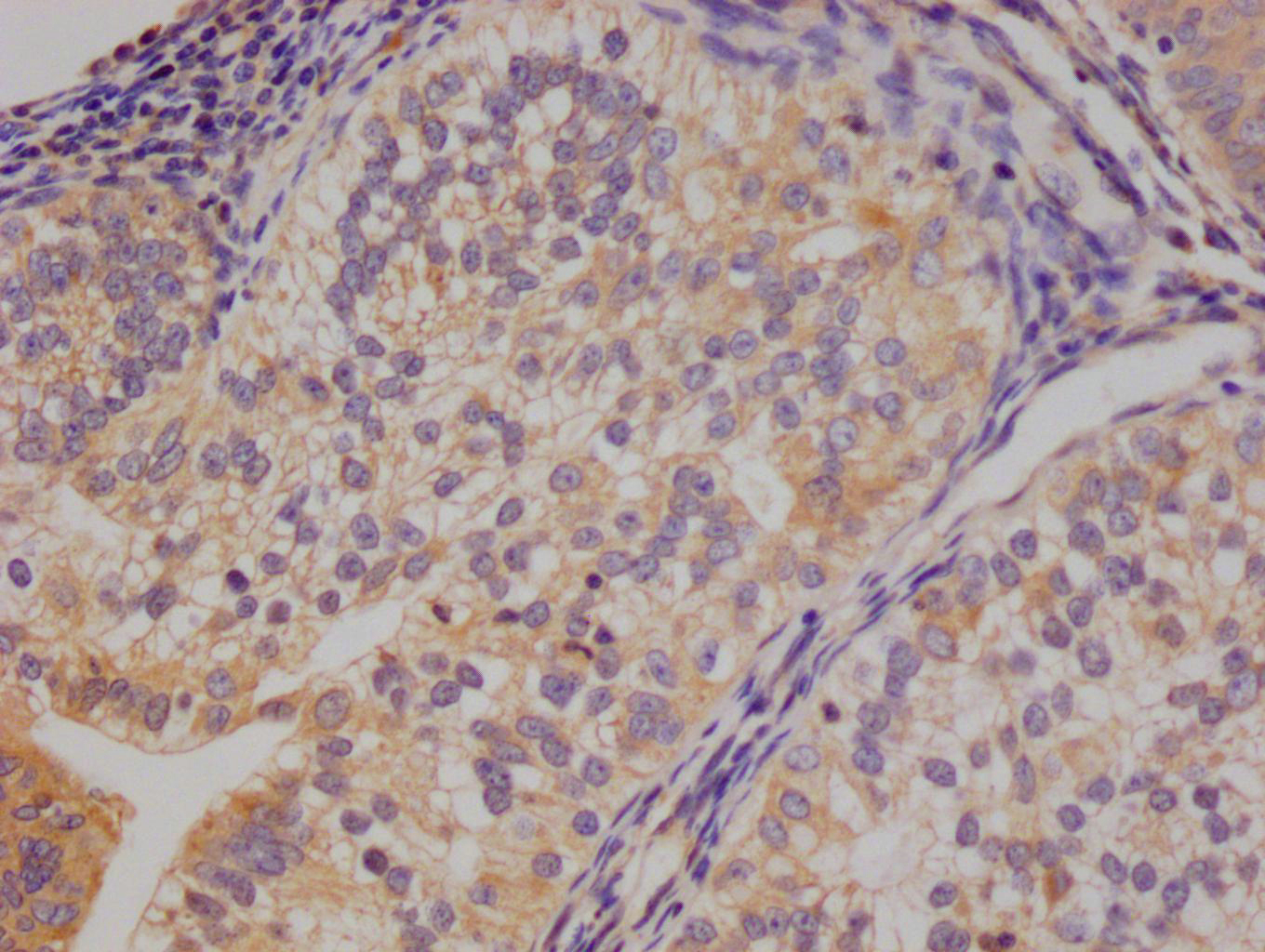
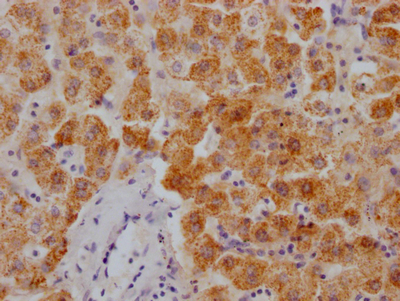
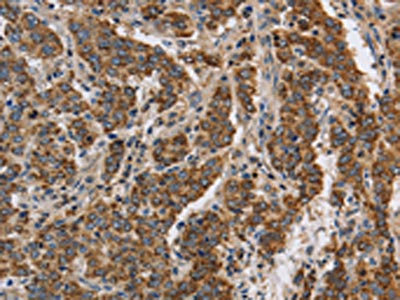 The image on the left is immunohistochemistry of paraffin-embedded Human liver cancer tissue using CSB-PA320879(KCNQ4 Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200)
The image on the left is immunohistochemistry of paraffin-embedded Human liver cancer tissue using CSB-PA320879(KCNQ4 Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200) -
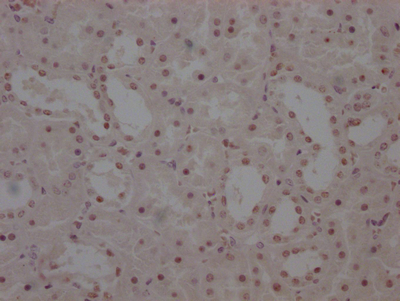
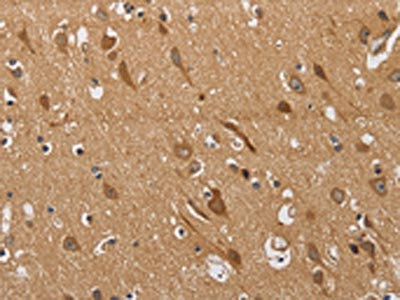 The image on the left is immunohistochemistry of paraffin-embedded Human brain tissue using CSB-PA320879(KCNQ4 Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200)
The image on the left is immunohistochemistry of paraffin-embedded Human brain tissue using CSB-PA320879(KCNQ4 Antibody) at dilution 1/40, on the right is treated with synthetic peptide. (Original magnification: ×200) -
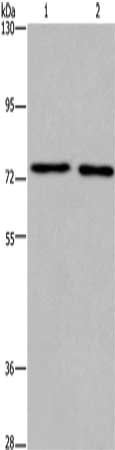 Gel: 6%SDS-PAGE, Lysate: 40 μg, Lane 1-2: Human fetal brain tissue, mouse brain tissue, Primary antibody: CSB-PA320879(KCNQ4 Antibody) at dilution 1/200, Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution, Exposure time: 7 minutes
Gel: 6%SDS-PAGE, Lysate: 40 μg, Lane 1-2: Human fetal brain tissue, mouse brain tissue, Primary antibody: CSB-PA320879(KCNQ4 Antibody) at dilution 1/200, Secondary antibody: Goat anti rabbit IgG at 1/8000 dilution, Exposure time: 7 minutes
-
-
其他:
产品详情
-
Uniprot No.:P56696
-
基因名:KCNQ4
-
别名:KCNQ4; Potassium voltage-gated channel subfamily KQT member 4; KQT-like 4; Potassium channel subunit alpha KvLQT4; Voltage-gated potassium channel subunit Kv7.4
-
宿主:Rabbit
-
反应种属:Human,Mouse
-
免疫原:Synthetic peptide of Human KCNQ4
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
-
抗体亚型:IgG
-
纯化方式:Antigen affinity purification
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:-20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol
-
产品提供形式:Liquid
-
应用范围:ELISA,WB,IHC
-
推荐稀释比:
Application Recommended Dilution ELISA 1:1000-1:2000 WB 1:200-1:1000 IHC 1:50-1:200 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Probably important in the regulation of neuronal excitability. May underlie a potassium current involved in regulating the excitability of sensory cells of the cochlea. KCNQ4 channels are blocked by linopirdin, XE991 and bepridil, whereas clofilium is without significant effect. Muscarinic agonist oxotremorine-M strongly suppress KCNQ4 current in CHO cells in which cloned KCNQ4 channels were coexpressed with M1 muscarinic receptors.
-
基因功能参考文献:
- KCNQ4 gene polymorphisms associated with susceptibility to noise-induced hearing loss. PMID: 29072670
- mutations in KCNQ4 gene are unlikely to be a major causative factor of ADNSHL in our studied patients from West Bengal, India, pointing to other genes might be responsible for ADNSHL in our studied patients PMID: 28802383
- The fundamental processes that dictate Kv7.4 activity. PMID: 27981364
- A novel KCNQ4 mutation, c.887 G > A (p.G296D), was identified in all five affected members in a Chinese family with autosomal dominant non-syndromic deafness 2. This mutation leads to a glycine-to-aspartic acid substitution at position 296 in the pore region of the KCNQ4 channel. PMID: 28340560
- Tannic acid activates Kv7.4 and Kv7.3/7.5 K(+) channels resulting in vasodilation. PMID: 26969140
- Gene and protein expression analyses show the predominance of KV7.4 channels over the other KV7 channel subtypes in human detrusor. PMID: 27761601
- Kv7.4 channels are present and functional in cardiac mitochondria; their activation exerts a significant cardioprotective role PMID: 26718475
- analysis of mechanistic insights into the critical roles of Ca(2+)/CaM regulation of the Kv7.4 channel under physiological and pathological conditions PMID: 26515070
- Interaction between G-protein betagamma subunits and Kv7.4 is crucial for channel responses to membrane voltage. PMID: 25941381
- genotype-phenotype correlation is analogous to that in KCNQ1 which causes autosomal dominant hereditary long QT syndrome 1 with milder phenotype and the autosomal recessive Jervell and Lange-Nielsen syndrome 1 with more severe phenotype PMID: 26036578
- This study provides evidence that mouse Kv7 channels may contribute differently to regulating the functional properties of cerebral and coronary arteries. PMID: 25476662
- The study identified a novel KCNQ4 mutation in a five generation Chinese family and a known KCNQ4 mutation in a six generation Chinese family. PMID: 25116015
- These findings suggest a protective role for Kv7.4 channels in the pulmonary circulation, limiting its reactivity to pressor agents and preventing hypoxia-induced pulmonary hypertension. PMID: 25361569
- Identified the c.211delC mutation in the KCNQ4 gene and the c.2967C>A (p.H989Q) mutation in the TECTA gene to be associated with high-frequency sensorineural hearing loss in a Japanese family. PMID: 24655070
- Kv7.4 currents are inhibited in a CB1 pathway repressed by endocannabinoid 2-AG PMID: 24927567
- In-frame deletion in KCNQ4 P-loop was identified in family members with autosomal dominant sensorineural hearing loss. PMID: 23443030
- Differential protein kinase C-dependent modulation of Kv7.4 and Kv7.5 subunits of vascular Kv7 channels. PMID: 24297175
- decreased cell surface expression and impaired conductance of the KCNQ4 channel are two mechanisms underlying hearing loss in DFNA2 PMID: 23750663
- A new typical audiogram configuration characterized by mid-frequency predominant hearing loss caused by the KCNQ4 V230E mutation. PMID: 23717403
- The present study supports the idea that a non-truncating mutation around the N-terminus of KCNQ4 pore helix may be related to moderate hearing loss. PMID: 23399560
- KCNQ4 surface expression was restored by HSP90beta in cells mimicking heterozygous conditions of the DFNA2 patients. PMID: 23431407
- The data of this study identified a dynamic redox sensor within neuronal M-channels, which mediates reciprocal regulation of channel activity by NO and ROS PMID: 23554485
- Data indicate a missense mutation encoding a Tyr270His located at the N-terminus of the highly conserved of KCNQ4 pore helix sequence. PMID: 22420747
- no sequence alterations that segregate with autosomal dominant non-syndromic deafness in either GJB3 or KCNQ4 PMID: 21651318
- This work describes a gene mutation that modulates touch sensitivity in mice and humans and establishes KCNQ4 as a specific molecular marker for rapidly adapting Meissner and a subset of hair follicle afferents. PMID: 22101641
- Data show that KCNQ4 and KCNE1 isoforms were suppressed in placentas from term preeclamptic women. PMID: 21730298
- Identified is novel mutation (c.664_681del) in KCNQ4 associated with hearing loss in a Korean family with dominantly inherited deafness. PMID: 20832469
- mutations in the pore region, namely L274H, W276S, L281S, G285C, and G296S, as well as the C-terminal mutant G321S in the heterologous expression system, yielded non-functional channels because of endoplasmic reticulum retention of the mutant channels PMID: 20966080
- Autosomal dominant progressive sensorineural hearing loss due to a novel mutation in the KCNQ4 gene. PMID: 21242547
- evidence of the cellular etiology and mechanisms of SGN degeneration in DFNA2. PMID: 20739290
- The W276S mutation has occurred three times independently, and most likely represents a hot spot for mutation in the KCNQ4 gene. PMID: 12112653
- DFNA2 locus was found to be related to hereditary sensorineural hearing loss. PMID: 12484650
- KCNQ3 interacts with KCNQ4. . A chimaera (KCNQ1-sid(Q3)) carrying the si domain of KCNQ3 within the KCNQ1 backbone interacted with KCNQ4. PMID: 12524525
- Src associates with KCNQ2-5 subunits but phosphorylates only KCNQ3-5. PMID: 15304482
- KCNQ4 phosphorylation via PKA and coupling to a complex that may include prestin can lead to the negative activation and the negative resting potential found in adult outer hair cells. PMID: 15660259
- Polymorphisms within the KCNQ4 gene are associated with susceptibility Noise-induced hearing loss. PMID: 16823764
- Results shows KCNQ4 SNPs were significantly associated with Age-related hearing impairment. PMID: 16917933
- a novel mutation of KCNQ4 gene was identified in patients with nonsyndromic deafness. PMID: 17033161
- In conclusion, this work demonstrates that inactivation is a key regulatory mechanism of Kv7.4 and Kv7.5 channels. PMID: 17237198
- high-resolution structure of the Kv7.4 A-domain Tail together with biochemical experiments show that the domain is a self-assembling, parallel, four-stranded coiled coil PMID: 17329207
- G296S mutant exerts a strong dominant-negative effect on potassium currents by reducing the wild type KCNQ4 channel expression at the cell surface PMID: 18030493
- among the allowed assembly conformations are KCNQ3/4 and KCNQ4/5 heteromers. PMID: 18786918
- KCNQ4 mutations associated with progressive sensorineural hearing loss. PMID: 18797286
- Two novel missense mutations and a stop mutation were detected in three American families predicted to have DFNA2-related deafness; The latter is the first DFNA2-causing stop mutation reported in KCNQ4. PMID: 18941426
显示更多
收起更多
-
相关疾病:Deafness, autosomal dominant, 2A (DFNA2A)
-
亚细胞定位:Basal cell membrane; Multi-pass membrane protein.
-
蛋白家族:Potassium channel family, KQT (TC 1.A.1.15) subfamily, Kv7.4/KCNQ4 sub-subfamily
-
组织特异性:Expressed in the outer, but not the inner, sensory hair cells of the cochlea. Slightly expressed in heart, brain and skeletal muscle.
-
数据库链接:
HGNC: 6298
OMIM: 600101
KEGG: hsa:9132
STRING: 9606.ENSP00000262916
UniGene: Hs.473058
Most popular with customers
-
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-