SCN1A Antibody
-
货号:CSB-PA044197
-
规格:¥1100
-
图片:
-
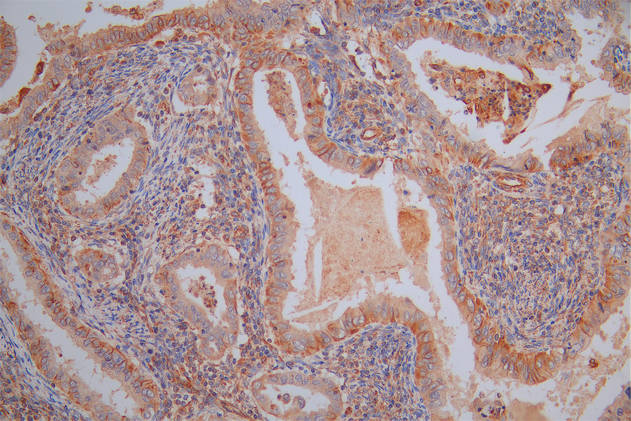
 The image on the left is immunohistochemistry of paraffin-embedded Human cervical cancer tissue using CSB-PA044197(SCN1A Antibody) at dilution 1/50, on the right is treated with synthetic peptide. (Original magnification: ×200)
The image on the left is immunohistochemistry of paraffin-embedded Human cervical cancer tissue using CSB-PA044197(SCN1A Antibody) at dilution 1/50, on the right is treated with synthetic peptide. (Original magnification: ×200) -
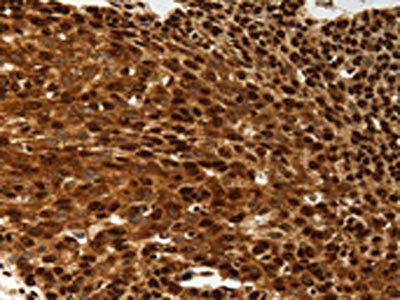
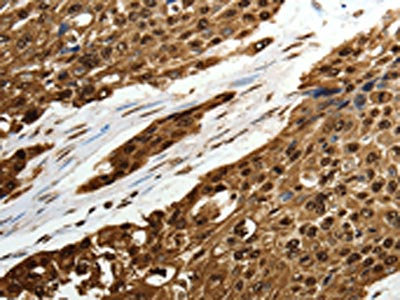 The image on the left is immunohistochemistry of paraffin-embedded Human esophagus cancer tissue using CSB-PA044197(SCN1A Antibody) at dilution 1/50, on the right is treated with synthetic peptide. (Original magnification: ×200)
The image on the left is immunohistochemistry of paraffin-embedded Human esophagus cancer tissue using CSB-PA044197(SCN1A Antibody) at dilution 1/50, on the right is treated with synthetic peptide. (Original magnification: ×200)
-
-
其他:
产品详情
-
Uniprot No.:P35498
-
基因名:
-
别名:brain sodium channel type I antibody; EIEE6 antibody; FEB3 antibody; FEB3A antibody; FHM3 antibody; GEFS+2 antibody; GEFSP2 antibody; HBSC I antibody; HBSCI antibody; NAC1 antibody; Nav 1.1 antibody; RBI antibody; SCN1 antibody; Scn1a antibody; SCN1A_HUMAN antibody; SMEI antibody; Sodium channel protein brain I alpha subunit antibody; Sodium channel protein brain I subunit alpha antibody; Sodium channel protein type 1 subunit alpha antibody; Sodium channel protein type I subunit alpha antibody; Sodium channel voltage gated type 1 alpha subunit antibody; Sodium channel voltage gated type I alpha polypeptide antibody; Voltage-gated sodium channel subunit alpha Nav1.1 antibody
-
宿主:Rabbit
-
反应种属:Human,Rat
-
免疫原:Synthetic peptide of Human SCN1A
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
-
抗体亚型:IgG
-
纯化方式:Antigen affinity purification
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:-20°C, pH7.4 PBS, 0.05% NaN3, 40% Glycerol
-
产品提供形式:Liquid
-
应用范围:ELISA,IHC
-
推荐稀释比:
Application Recommended Dilution ELISA 1:2000-1:5000 IHC 1:50-1:200 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Mediates the voltage-dependent sodium ion permeability of excitable membranes. Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient. Plays a key role in brain, probably by regulating the moment when neurotransmitters are released in neurons. Involved in sensory perception of mechanical pain: activation in somatosensory neurons induces pain without neurogenic inflammation and produces hypersensitivity to mechanical, but not thermal stimuli.
-
基因功能参考文献:
- Our cases represent a novel association between SCN1A and sudden infant death syndrome (SIDS), extending the SCN1A spectrum from epilepsy to SIDS. PMID: 29601086
- The effect of Ca(2+), domain-specificity, and CaMKII on CaM binding to NaV1.1 has been reported. PMID: 30142967
- This first genetic study of Dravet syndrome in Africa confirms that de novo SCN1A variants underlie disease in the majority of South African patients. PMID: 30321769
- these data suggest that MDH2, functioning as an RNA-binding protein, is involved in the posttranscriptional downregulation of SCN1A expression under seizure condition. PMID: 28433711
- Postzygotic mutation is a common phenomenon in SCN1A-related epilepsies. Participants with mosaicism have on average milder phenotypes, suggesting that mosaicism can be a major modifier of SCN1A-related diseases. PMID: 29460957
- SCN1A and SCN2A mutations and clinical/EEG features in Chinese patients from epilepsy or severe seizures PMID: 29649454
- rs7668282 (UGT2B7, T>C) was more prevalent in sodium valproate (VPA)-resistant patients than drug-responsive patients. rs2242480 (CYP3A4, C>T) and rs10188577 (SCN1A, T>C) were more prevalent in drug-responsive patients compared to drug-resistant patients. In children with generalized seizures on VPA therapy, polymorphisms of UGT2B7, CYP3A4, and SCN1A genes were associated with seizure reduction. PMID: 29679912
- Meta-analysis indicates that the A-allele of SCN1A rs2298771 polymorphism, especially AA genotype, may play an important role in responsiveness to sodium channel blocking antiepileptic drugs, while SCN1A rs3812718 polymorphism is not associated. PMID: 29582177
- Study established induced pluripotent stem cells (iPSC)from two Dravet syndrome patients with different mutations in SCN1A and subsequently differentiated them into forebrain GABAergic neurons. Unique genetic alterations of SCN1A differentially impacted electrophysiological impairment of the neurons, and the impairment's extent corresponded with the symptomatic severity of the donor from which the iPSCs were derived. PMID: 29295803
- miR155 may be associated with the risk of seizure and SCN1A may be a target gene of miR155. PMID: 29115566
- we report a Chinese familial hemiplegic migraine type 3 family with a novel mutation in the SCN1A gene. PMID: 27919014
- The polymorphic SCN1A c.3184A>G/p.Thr1067Ala G allele was associated with a lower risk of epilepsy and a higher remission rate in Slovenian children and adolescents with epilepsy. PMID: 28753467
- The evaluation of an SCN1A mutation. PMID: 27582020
- These findings provide insight into a pharmacophore on domain IV voltage sensor through which modulation of inactivation gating can inhibit or facilitate Nav1.1 function. PMID: 28607094
- SCN1A rs3812718 polymorphism is a risk factor for GEFS+, and the population carrying T allele may have an increased risk of GEFS. PMID: 29429462
- study showed a significant association between rs6730344, rs6732655 and rs10167228 polymorphisms in the intronic regions of the SCN1A gene and refractory epilepsy, thus emerging as risk factors for drug resistance PMID: 29353705
- showed large-scale developmental brain changes in patients with epilepsy and SCN1A gene mutation, which may be associated with the core symptoms of the patients PMID: 28664031
- These data provide evidence that Nav1.1 is indeed vulnerable to deltamethrin modification at lower concentrations than Nav1.6, and this effect is primarily mediated during the resting state. PMID: 28007400
- SCN1A deletions are more common than SCN1A duplications among Dravet syndrome patients, and the most common types are whole SCN1A deletions. PMID: 29188601
- Two novel missense mutations: p.G325A in the KDM6A gene responsible for Kabuki syndrome and p.G1877V in the SCN1A gene responsible for generalized epilepsy with febrile seizures plus were identified using the TruSight One sequencing panel. PMID: 28442529
- Similar to the known FHM3 mutations, this novel mutation predicts hyperexcitability of GABAergic inhibitory neurons. PMID: 26763045
- Study found in patients with focal seizures a seemingly uneven distribution of mutations within the SCN1A gene. Study identified valproic acid, stiripentol, and clobazam to be 3 medications that have the best effect on management of focal seizures due to an SCN1A mutation similar to what is seen in most cases of Dravet Syndrome. PMID: 27777328
- A novel SCN1A mutation was found in an 8-year-old boy with GEFS+. The father has the same mutation, but he only had childhood simple febrile seizures. PMID: 28262406
- In this study, we performed exome sequencing in six patients with SCN1A-negative Dravet syndrome to identify other genes related to this disorder..the data in this study identify GABRB3 as a candidate gene for Dravet syndrome PMID: 28544625
- Dravet syndrome-derived inhibitory neurons showed deficits in sodium currents and action potential firing, which were rescued by a Nav1.1 transgene, whereas Dravet excitatory neurons were normal. PMID: 27458797
- The results further substantiate the contribution of SCN1A in response and therapy optimization with PHT monotherapy. PMID: 27245092
- Rather than a single common gene/variant modifying clinical outcome in SCN1A-related epilepsies, our results point to the cumulative effect of rare variants with little to no measurable phenotypic effect (i.e., typical genetic background) unless present in combination with a disease-causing truncation mutation in SCN1A. PMID: 28686619
- The frequency of pathogenic variants was 4.17% in the patients with refractory epilepsy and Dravet syndrome and 11.1% in DS patients. L1775P missense mutation was predicted to be damaging with a score of 100% by the PolyPhen-2 program. PMID: 28525652
- genetic variants in 3'UTR of SCN1A PMID: 27473590
- Among these transmissions were two likely disease-causing mutations: an SCN1A mutation transmitted to an SUDC proband and her sibling with Dravet syndrome, as well as an SLC6A1 mutation in a proband with epileptic encephalopathy. PMID: 26716362
- study presents a phenotype-genotype correlation for SCN1A; described a distinct SCN1A phenotype, early infantile SCN1A encephalopathy, which is readily distinguishable from the Dravet syndrome and genetic epilepsy with febrile seizures plus PMID: 28794249
- This study demonstrated that early-life prolonged FSs have a profound long-term impact on neuronal function and adult seizure phenotypes in a mouse model of human SCN1A dysfunction. PMID: 28373025
- Pathogenic, likely pathogenic, and benign variants in SCNs were identified using databases of sodium channel variants. Benign variants were also identified from population-based databases. Eight algorithms commonly used to predict pathogenicity were compared. In addition, logistic regression was used to determine if a combination of algorithms could better predict pathogenicity. PMID: 28518218
- this study showed that SCN1A testing be considered in all individuals with febrile seizures or Dravet syndrome , as well as in familial cases consistent with febrile seizures. PMID: 28084635
- This study found significant differences in the distribution of truncating and missense variants across the SCN1A sequence among healthy individuals, patients with Dravet syndrome. PMID: 28012175
- A heterozygous mutation h1u-1962 T > G was identified in a patient with partial epilepsy and febrile seizures, which was aggravated by oxcarbazepine. PMID: 26969601
- SCN1A mutations may alter axonal function, causing motor neuropathy/neuronopathy. This may contribute to gait disturbance and orthopedic misalignment, which is characteristic of patients with Dravet syndrome. PMID: 27316242
- The association study indicated that age at first seizure and frameshift mutations of SCN1A were associated with Dravet syndrome. PMID: 28202706
- Study reported the range of rare copy number variants found in SCN1A gene in a series of Welsh patients with childhood-onset epilepsy and intellectual disability and identified clearly or likely pathogenic CNVs in 8.8 % of the patients including 5 rare de novo deletions. PMID: 27113213
- Our findings suggest that SCN1A mutation leads to changes in the dopamine system that may contribute to the behavioral abnormalities in DS. PMID: 26841829
- This study demonstrated that a significant higher frequency of the AG genotype (p=0.001) and G allele (p=0.006) of SCN1A polymorphism in epileptic patients than in controls. PMID: 27498208
- Dravet syndrome is associated with mutations in the sodium channel alpha1 subunit gene (SCN1A) in 70-80% of individuals. PMID: 27264139
- The presence of SCN1A mutations and absence of mutations in ATP1A2 or CACNA1A suggest that the Polish patients represent FHM type 3. PMID: 26747084
- The findings of this study in scn1a mutant zebrafish suggest that glucose and mitochondrial hypometabolism contribute to the pathophysiology of DS. PMID: 27066534
- Mutations in SCN1A, the gene that encodes the alpha subunit of voltage-gated sodium channel Nav1.1, can cause epilepsies. PMID: 26731440
- Retrospective study to survey the efficacy of antiepileptic drugs in Dravet syndrome with different SCN1A genotypes PMID: 26183863
- No causative variants were identified in any of non-DS epileptic patients in our cohort, suggesting a minor, but not irrelevant role for SCN1A in patients with other types of childhood epilepsy. PMID: 27045673
- Mutations in SCN1A and SCN2A are a predisposing factor of acute encephalopathy with biphasic seizures and late reduced diffusion PMID: 26311622
- Evaluation of Presumably Disease Causing SCN1A Variants in a Cohort of Common Epilepsy Syndromes. PMID: 26990884
- The SCN1A IVS5-91G>A SNP is associated with susceptibility to epilepsy. SNPs in EPHX1 gene are influencing CBZ metabolism and disposition PMID: 26555147
显示更多
收起更多
-
相关疾病:Generalized epilepsy with febrile seizures plus 2 (GEFS+2); Epileptic encephalopathy, early infantile, 6 (EIEE6); Intractable childhood epilepsy with generalized tonic-clonic seizures (ICEGTC); Migraine, familial hemiplegic, 3 (FHM3); Febrile seizures, familial, 3A (FEB3A)
-
亚细胞定位:Cell membrane; Multi-pass membrane protein.
-
蛋白家族:Sodium channel (TC 1.A.1.10) family, Nav1.1/SCN1A subfamily
-
数据库链接:
HGNC: 10585
OMIM: 182389
KEGG: hsa:6323
STRING: 9606.ENSP00000303540
UniGene: Hs.22654
Most popular with customers
-
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-
-