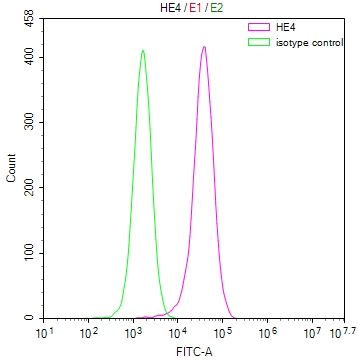
SCN2A Antibody
-
货号:CSB-PA040291
-
规格:¥880
-
其他:
产品详情
-
Uniprot No.:Q99250
-
基因名:
-
别名:HBSC II antibody; NAC2 antibody; Scn2a antibody; SCN2A_HUMAN antibody; SCN2A1 antibody; SCN2A2 antibody; Sodium channel protein brain II subunit alpha antibody; Sodium channel protein type 2 subunit alpha antibody; Sodium channel protein type II subunit alpha antibody; Sodium channel protein, brain II subunit alpha antibody; Voltage gated sodium channel subunit alpha Nav1.2 antibody; Voltage-gated sodium channel subunit alpha Nav1.2 antibody
-
宿主:Rabbit
-
反应种属:Human,Mouse,Rat
-
免疫原:Synthesized peptide derived from the Internal region of Human Na+ CP type IIα.
-
免疫原种属:Homo sapiens (Human)
-
标记方式:Non-conjugated
-
抗体亚型:IgG
-
纯化方式:The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
-
浓度:It differs from different batches. Please contact us to confirm it.
-
保存缓冲液:Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
-
产品提供形式:Liquid
-
应用范围:IHC, ELISA
-
推荐稀释比:
Application Recommended Dilution IHC 1:100-1:300 ELISA 1:5000 -
Protocols:
-
储存条件:Upon receipt, store at -20°C or -80°C. Avoid repeated freeze.
-
货期:Basically, we can dispatch the products out in 1-3 working days after receiving your orders. Delivery time maybe differs from different purchasing way or location, please kindly consult your local distributors for specific delivery time.
产品评价

相关产品
靶点详情
-
功能:Mediates the voltage-dependent sodium ion permeability of excitable membranes. Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient. Implicated in the regulation of hippocampal replay occurring within sharp wave ripples (SPW-R) important for memory.
-
基因功能参考文献:
- Review: Dysfunction in SCN2A has been recently recognized as a major cause of neurodevelopmental disorders (NDDs), including epilepsy, intellectual disability (ID), and autism spectrum disorder (ASD). Loss of NaV1.2 function contributes to ASD and ID, whereas gain of function contributes to early onset epilepsy. Sodium channel function can be enhanced or suppressed using pharmacology. PMID: 29691040
- We utilized a targeted next generation sequencing (NGS) approach on a girl with early-onset seizures and Rett-like features, including autistic behavior, limited hand function with chorea, and profound intellectual disability, to identify novel missense mutation (c.1270G>A; p.V424M) in the SCN2A gene, which encodes the alphaII-subunit of the voltage-gated Na+ channel (Nav1.2). PMID: 28709814
- SCN2A autism spectrum disorder (ASD)-associated variants dampened or eliminated channel function in transfected HEK293 cells. A compartmental model of developing excitatory neurons demonstrated that all ASD variants, regardless of their mechanism of action, resulted in deficits in neuronal excitability. Corresponding analysis of mature neurons predicted minimal change in neuronal excitability. PMID: 28256214
- clinical and experimental data suggest a correlation between age at disease onset, response to sodium channel blockers and the functional properties of mutations in children with SCN2A-related epilepsy. PMID: 28379373
- The most frequently mutated gene in early-onset seizure and severe developmental delay disorders was SCN2A (11 patients, 3%), occurring de novo in all patients. PMID: 26993267
- Data indicate eight new cases with overlapping duplications at 2q24 and SCN3A was not involved in duplication, suggesting that having an extra copy of SCN2A has an effect on epilepsy pathogenesis. PMID: 27153334
- This study demonstrated the association of SCN2A rs1864885 A > G, in Japanese childhood encephalopathy patients. PMID: 27538648
- Our findings broaden the clinical spectrum observed with SCN2A gain-of-function mutations, showing that fairly different biophysical mechanisms can cause a convergent clinical phenotype of neonatal seizures and later onset episodic ataxia. PMID: 26645390
- We argue that very rare, loss-of-function mutations at SCN2A act in a moderately penetrant manner to increase the risk of developing several neuropsychiatric disorders including seizure disorders, intellectual disability, autism and schizophrenia. PMID: 26555645
- Findings provide convergent evidence that a common polymorphism in SCN2A accounts for significant interindividual variability in human general cognitive ability, possibly by modulating prefrontal cortex physiology PMID: 25961639
- This study provide the evidence SCN2A mutation releate to Autism Spectrum Disorder. PMID: 26637798
- A heterozygous mutation (c.3631G > A; p.E1211K) was identified in exon 21 of SCN2A gene. This is the first case of SCN2A mutation identified in Chinese. PMID: 25459969
- Case suggests that SCN2A mutations might predispose children to repetitive encephalopathy with variable clinical and imaging findings. PMID: 25457084
- SCN2A is the second most common cause of epilepsy of infancy with migrating focal seizures PMID: 26291284
- Two lysine residues (residues 105-106)of AnkG are critical for Nav1.2 but not KCNQ3 channel binding. PMID: 25998125
- Epileptic encephalopathy related to mutations in the SCN2A genes. PMID: 25818041
- the mechanism of AED resistance remains uncertain, which led us to evaluate the impact of polymorphisms of the SCN1A, SCN2A and ABCC2 genes on the AED response in Chinese Han patients with epilepsy in the present study. PMID: 25155934
- mutation of SCN2A is associated with a range of epilepsy phenotypes including severe infantile-onset epilepsy PMID: 24659627
- identified common variants in SCN2A that, in the context of schizophrenia and risk for schizophrenia, show substantial and consistent associations with broad cognitive performance, brain physiology, and mRNA expression in the brain. PMID: 24718902
- Following SCN2A knockdown, the concentration of Cu-Zn SOD declined and the si-SCN2A vector group showed a repeated discharge. PMID: 24220630
- Exome sequencing reveals a de novo mutation in SCN2A linked to a genetically heterogeneous disorder with epilepsy and intellectual disability, expanding the phenotype of SCN2A mutations. PMID: 24579881
- An SCN2A missense mutation is associated with infantile spasms and bitemporal hypometabolism. PMID: 23827426
- Mutation of SCN2A induces neuronal hyperexcitability, resulting in infantile epilepsy with favorable outcome PMID: 23758435
- SCN2A mutations are an important genetic cause of Ohtahara syndrome. Given the wide clinical spectrum associated with SCN2A mutations, genetic testing for SCN2A should be considered for children with different epileptic conditions. PMID: 23935176
- study identified a de novo SCN2A mutation as the etiology for Ohtahara syndrome in monozygotic twins associated with a unique dentate-olivary dysplasia in the deceased twin PMID: 23550958
- Identification of a novel SCN2A mutation in a family with infantile seizures with onset between 6 and 8 months provides further confirmation that this gene is involved in families with a delayed age of onset. PMID: 23360469
- This finding suggests that SCN2A mutation is a predisposing factor for acute encephalopathy. PMID: 22591750
- This study demonistrated that associated with a duplication of the SCN2A and SCN3A gene cluster on 2q24 in patient with early onset epilepsy. PMID: 23016767
- This study demonistrated the detailed functional analysis of Na(v)1.2 mutant, R1312T, which was originally found in a child with Dravet syndrome. PMID: 22677033
- Functional studies of SCN2A mutations show that they can cause divergent biophysical defects in Na(V)1.2 and impair cell surface expressions[review] PMID: 22029951
- The results of this study shown that both A1685V (GEFS+) and A1685D (SMEI) mutant Nav1.1 channels are characterized by complete loss of function when they are expressed alone PMID: 22525008
- association of the gene SCN2A, previously identified in epilepsy syndromes, with the risk of autism PMID: 22495306
- Genetic variants in the voltage-gated ion channel SCN2A dramatically influence the phenotype of mice carrying an SCN1A mutation as well as the seizures caused by the SCN1A mutation. PMID: 21156207
- Environmental enrichment from birth reduces spontaneous seizures and neuronal damage in the Q54 model of temporal lobe epilepsy. PMID: 21762452
- Thermodynamic and structural studies of Calmodulin (CaM)-Na(v)1.2(IQp) interactions show that apo and (Ca(2+))(4)-CaM adopt distinct conformations that both permit tight association with Na(v)1.2(IQp) during gating. PMID: 21439835
- Deletions in SCN2A gene is associated with autistic features and developmental delay. PMID: 20346423
- Transient expression of seizures occur due to a gain-of-function of mutant Na(V)1.2 channel. PMID: 20371507
- in autism families, the variant R1902C in SCN2A is located in the calmodulin binding site and was found to reduce binding affinity for calcium-bound calmodulin. PMID: 12610651
- We found a nonsense mutation of SCN2A in a patient with intractable epilepsy and severe mental decline. The phenotype is like severe myoclonic epilepsy in infancy but distinct because of partial epilepsy, delayed onset and no temperature sensitivity PMID: 15028761
- Calmodulin mediates Ca2+ sensitivity of Nav1.2 and Nav1.5 sodium channels PMID: 15316014
- Although data suggest that SCN2A1 activity does not directly influence membrane potential, intracellular Ca(2+) release, or proliferation in normal human pulmonary artery smooth muscle cells, its physiological functions remain unresolved. PMID: 16052353
- Genetic interaction between the combined mild alleles of monogenic epilepsy genes KCNQ2 and SCN2A1 results in severe epilepsy in transgenic mice. PMID: 16464983
- Data show that the varied effects of beta1 and beta2 on Nav1.5 and Nav1.2 gating are apparently synergistic and highlight the complex manner, through subunit- and sugar-dependent mechanisms, by which Nav activity is modulated. PMID: 16847056
- The SCN2A gene was tested for a possible role in hippocampal abnormalities in familial mesial temporal lobe epilepsy. We conclusively ruled out the SCN2A gene as a candidate in FMTLE. PMID: 16914293
- two large families with benign familial neonatal-infantile seizure (BFNIS) and novel SCN2A mutations; the families had 12 & 9 affected individuals, respectively, with phenotypes consistent with BFNIS; two mutations were discovered in SCN2A (E430Q; I1596S) PMID: 17386050
- analysis of neonatal & adult splice forms of NaV1.2 with a benign familial neonatal-infantile seizures mutation; developmentally regulated NaV1.2 splicing may be one mechanism that counters the normally high excitability of neonatal neurons PMID: 17467289
- Characterization of 5' untranslated regions SCN2A, and identification of cis-conserved noncoding sequences PMID: 17544618
- In south china, the R188W mutation of the SCN2A gene not releate to children with febrile seizures. PMID: 17641256
- Allele frequencies of both the D2S111 and the D2S124 polymorphisms of the SCN2A gene were not significantly different between cases with a specific idiopathic generalized epilepsy subtype (with generalized tonic-clonic seizures) and healthy controls. PMID: 17715289
- Nav1.2 immunostaining was not observed along demyelinated axons in chronic lesions but was expressed by scar and reactive astrocytes within the plaque. PMID: 17805013
显示更多
收起更多
-
相关疾病:Seizures, benign familial infantile, 3 (BFIS3); Epileptic encephalopathy, early infantile, 11 (EIEE11)
-
亚细胞定位:Cell membrane; Multi-pass membrane protein.
-
蛋白家族:Sodium channel (TC 1.A.1.10) family, Nav1.2/SCN2A subfamily
-
数据库链接:
HGNC: 10588
OMIM: 182390
KEGG: hsa:6326
STRING: 9606.ENSP00000283256
UniGene: Hs.93485
Most popular with customers
-
-
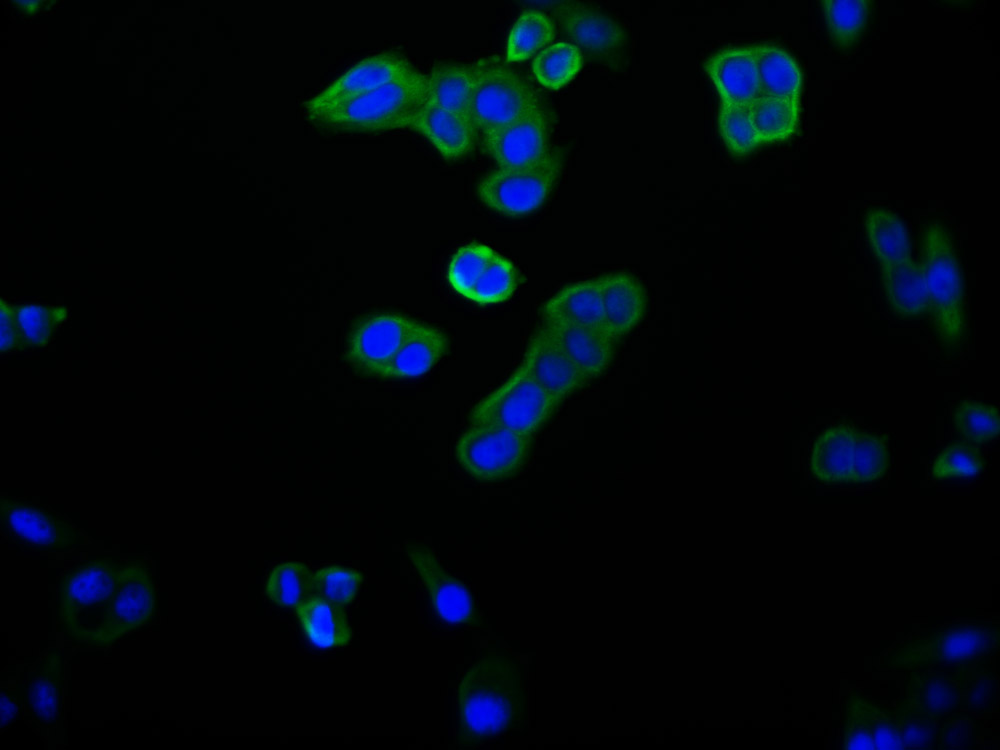
YWHAB Recombinant Monoclonal Antibody
Applications: ELISA, WB, IF, FC
Species Reactivity: Human, Mouse, Rat
-
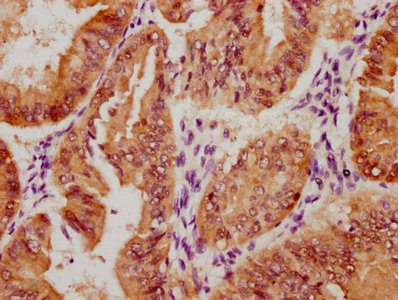
Phospho-YAP1 (S127) Recombinant Monoclonal Antibody
Applications: ELISA, WB, IHC
Species Reactivity: Human
-
-
-
-
-