组蛋白甲基化
1999年首次描述了组蛋白甲基化与DNA转录之间的联系,但与组蛋白乙酰化不同的是,随后很快就鉴定出了特定的组蛋白甲基转移酶(HMTs)[1,2]。
1. 什么是组蛋白甲基化?
组蛋白甲基化是发生在H3和H4组蛋白的N-末端的赖氨酸(K)或精氨酸(R)残基上的甲基化。组蛋白精氨酸残基可以是单甲基化(me1),不对称或对称二甲基化(me2as或me2s),而赖氨酸残基可以经历单甲基化,二甲基化(me2)和三甲基化(me3)。
在哺乳动物细胞中,有六个广泛研究的甲基化位点,包括H3K4,H3K9,H3K27,H3K36,H3K79和H4K20。至于精氨酸甲基化,主要发生在H3R2,H3R8,H3R17,H3R26和H4R3 [3]。与通过减弱组蛋白-DNA相互作用引起转录激活的组蛋白乙酰化不同,组蛋白甲基化增加了组蛋白的碱性和疏水性,并影响DNA-转录因子的相互作用,从而激活或抑制基因表达。
2. 参与组蛋白甲基化的三个关键组分
组蛋白甲基化是通过三个关键组分的作用完成的,包括编码酶、读取酶和擦除酶。编码酶,也称为组蛋白甲基转移酶(HMTs),从S-腺苷甲硫氨酸(AdoMet)向组蛋白尾部转移甲基基团。读取酶识别和结合甲基基团,最终影响基因表达。擦除酶,也称为组蛋白去甲基酶(HDMs),去除甲基基团。HMTs和HDMs共同维持适当的全局组蛋白甲基化水平,从而调控基因表达模式。
2.1 组蛋白甲基转移酶
组蛋白甲基转移酶(HMTs)根据其结构和修饰位置分为两类:组蛋白赖氨酸甲基转移酶(KMTs)和蛋白精氨酸甲基转移酶(PRMTs)。不同的酶负责不同位点的甲基化。
基于它们的细胞定位,HAT可分为两大类:A型HATs位于细胞核,乙酰化核小体组蛋白和其他染色质相关蛋白,而B型HATs位于细胞质,专门乙酰化新合成的游离组蛋白,对转录没有直接影响。
KMTs通过将甲基基团从S-腺苷甲硫氨酸转移至组蛋白赖氨酸残基的ε-氮,对组蛋白赖氨酸残基进行单甲基化,二甲基化或三甲基化。根据催化结构域序列,KMTs分为两个家族:含SET结构域的KMTs和不含SET结构域的KMTs。
PRMTs通过对组蛋白精氨酸残基的鸟嘌呤基团进行单甲基化或二甲基化来实现。它们主要分为类型I和类型II酶,启动形成单甲基化(MMA)中间体。然后,由类型I PRMTs(PRMT 1-4,6,8和9)产生精氨酸残基的不对称二甲基化(aDMA)和由类型II PRMTs(PRMT5和7)产生的对称二甲基化(sDMA)。
| 组蛋白甲基转移酶(HMT) | 亚型 | 甲基化位点 | 甲基化位点 | 染色质状 |
|---|---|---|---|---|
| KMTs | SET domain-containing KTMs | H3K4 | SET1A/KMT2F | 活跃 |
| SET1B/KMT2G | ||||
| MLL1/KMT2A | ||||
| MLL2/KMT2B | ||||
| MLL3/KMT2C | ||||
| MLL4/KMT2D | ||||
| SMYD1/KMT3D | ||||
| SMYD2/KMT3C | ||||
| SET7/9/KMT7 | ||||
| PRDM9 | ||||
| H3K9 | SUV39H1/KMT1A | 不活跃 | ||
| SUV39H2/KMT1B | ||||
| G9a/KMT1C | ||||
| GLP/KMT1D | ||||
| SETDB1/KMT1E | ||||
| PRDM family: (PRDM1, PRDM4, PRDM5, PRDM14) | ||||
| H3K27 | EZH1/KMT6B | 不活跃 | ||
| EZH2/KMT6A | ||||
| H3K36 | SETD2/KMT3A | 活跃 | ||
| NSD1/KMT3B | ||||
| NSD2/KMT3G | ||||
| NSD3/KMT3F | ||||
| SMYD2/KMT3C | ||||
| ASH1L/KMT2H | ||||
| SETD3 | ||||
| SETMAR | ||||
| H4K20 | SET8/KMT5A | 不活跃 | ||
| SUV4-20H1/KMT5B | ||||
| SUV4-20H2/KMT5C | ||||
| Non-SET domain-containing KTMs | H3K79 | DOT1L/KMT4 | 活跃 | |
| PRMT family | H3R2 | PRMT5 | 活跃 | |
| PRMT6 | 不活跃 | |||
| PRMT7 | 活跃 | |||
| H3R8 | PRMT2 | 不活跃 | ||
| PRMT5 | ||||
| H3R17 | PRMT4 | 活跃 | ||
| H3R26 | PRMT4 | 活跃 | ||
| H4R3 | PRMT5 | 活跃 | ||
| PRMT7 | 不活跃 | |||
2.2 组蛋白去甲基酶
2004年首次发现的LSD1是一种特异性去甲基化组蛋白H3K4的酶,随后鉴定出了几种在转录调控中发挥作用的去甲基酶。去甲基酶的发现纠正了组蛋白甲基化是可逆的观念。有两个主要的KDM家族:KDM1家族和含有Jumonji C(JmjC)结构域的家族。
| 分类 | 组蛋白去甲基化酶 | 底物特异性 | 功能 |
|---|---|---|---|
| KDM1 family | KDM1A/LSD1 | H3K4me1/2, H3K9me1/2 | 转录激活或抑制,异染色质形成 |
| KDM1B/LSD2 | H3K4me1/2 | 转录抑制 | |
| JmjC family | KDM2A | H3K36me1/2 | 转录延伸 |
| KDM2B | H3K36me1/2 | 转录延伸 | |
| KDM3A | H3K9me1/2 | 雄激素受体基因激活,精子发生 | |
| KDM3B | H3K9me1/2 | 转录激活 | |
| KDM4A | H3K9me2/3, H3K36me2/3 | 转录抑制,基因组完整性 | |
| KDM4B | H3K9me2/3, H3K36me2/3 | 异染色质形成 | |
| KDM4C | H3K9me2/3, H3K36me2/3 | 可能的致癌基因 | |
| KDM4D | H3K9me2/3, H3K36me2/3 | 转录激活或抑制 | |
| KDM5A | H3K4me2/3 | 视网膜母细胞瘤相互作用蛋白 | |
| KDM5B | H3K4me1/2/3 | 转录抑制 | |
| KDM5C | H3K4me2/3 | X连锁智力障碍 | |
| KDM5D | H3K4me2/3 | 男性特异性抗原 | |
| KDM6A | H3K27me2/3 | 转录激活 | |
| KDM6B | H3K27me2/3 | 转录激活 | |
| KDM7A | H3K9me1/2, H3K27me1/2 | 转录激活 | |
| KDM7B | H3K9me1/2 | 转录激活 | |
| NO66 | H3K4me2/3, H3K36me2/3 | 转录抑制 |
2.3 读取蛋白
组蛋白甲基化的读者蛋白通过甲基化赖氨酸结合结构域(包括植物家庭结构域(PHD)、染色体结构域(chromo)、图多尔(tudor)、脯氨酸-色氨酸-色氨酸-脯氨酸(PWWP)、WD40、BAH、ADD、ankyrin重复、MBT 和 zn-CW结构域)来识别甲基化的组蛋白。此外,这些蛋白质可以通过评估它们的甲基化状态和相邻氨基酸序列,来甄别受甲基化的赖氨酸目标。
用于组蛋白甲基化的不同类型的读者结构域是根据它们识别不同甲基化位点的能力进行分类的。PHD手指结构域负责识别H3K4甲基化,而染色体结构域对K9或K27甲基化具有特异性;这些染色体结构域亚家族分别适用于各自的位点。此外,PWWP结构域与K36甲基化的识别相关。
| PTMs | 位置 | 结构域 | 读取者 |
|---|---|---|---|
| Lysine Methylation | H3K4 | Chromo | CHD1 |
| PHD | RAG2, ING2, BPTF, TAF3, PHF2, ING4, YNG1, PHF8, BHC80, AIRE | ||
| Tudor | JMJD2A, JMJD2C, Sgf29 | ||
| WD40 | WDR5, WDR9 | ||
| ADD | Dnmt3L | ||
| MBT | PHF20L1 | ||
| Zf-CW | ZCWPW1 | ||
| H3K9 | Chromo | HP1, CDY1, CDYL, CDYL2 | |
| PHD | SMCX | ||
| Tudor | TDRD7, UHRF1 | ||
| WD40 | EED, LRWD1 | ||
| Ankyrin Repeats | G9a/GLP | ||
| H3K27 |
Chromo | PC, CDYL, CDYL2, CBX7, MPP8 | |
| WD40 | EED, LRWD1 | ||
| H3K36 |
PWWP | DNMT3A, BRPF1, NSD1, NSD2, NSD3, MSH-6, N-PAC | |
| Chromo | Eaf3, MSL3, MRG15 | ||
| H3K79 | Tudor | 53BP1 | |
| H4K20 |
Tudor | 53BP1/Crb2, PHF20 | |
| MBT | PHF20L1, L3MBTL1,Sfmbt1 | ||
| WD40 | LRWD1 | ||
| PWWP | Pdp1 | ||
| H1K26 | MBT | L3MBTL1 | |
| WD40 | EED | ||
| Arginine Methylation | H3R17 | Tudor | TDRD3 |
| H4R3 | Tudor | TDRD3 | |
| ADD | Dnmt3a |
表格信息来源: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3131977/
3. 组蛋白甲基化的机制
组蛋白甲基化通过吸引各种转录因子而不是直接修改染色质结构,从而控制基因表达。当组蛋白甲基转移酶(HMTs)向H3和H4上的赖氨酸或精氨酸残基添加甲基基团,与活跃染色质相关联时,转录激活因子(TA)被吸引到这些位点,促进基因表达。
另一方面,当HMTs靶向与染色质抑制相关的H3和H4上的不同残基时,甲基化区域可以吸引转录抑制因子(TR),导致基因沉默。组蛋白甲基化的转录影响可以被组蛋白去甲基酶(HDMs)逆转。维持适当的基因表达依赖于HMT和HDM酶的平衡活动。
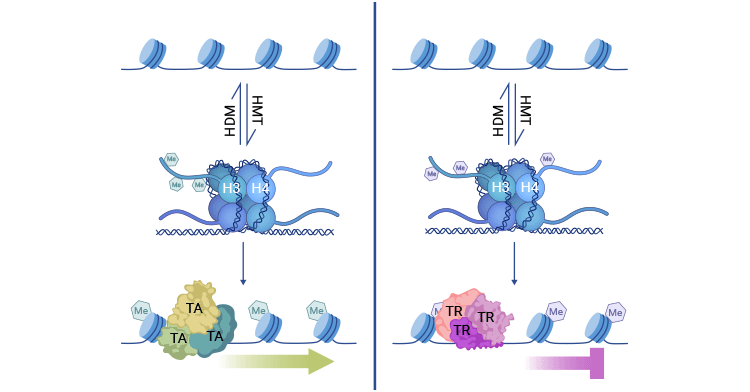
图1. 组蛋白甲基化机制
图片参考来源: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7820808/
4. 组蛋白甲基化的功能
组蛋白甲基化是一个动态且可逆的过程,在各种生物事件中发挥着关键作用,包括异染色质形成、X染色体失活、基因组稳定性、转录调控、细胞生长、新陈代谢、信号传导和DNA损伤应答(DDR)[4,5]。
组蛋白甲基化可以激活或抑制基因表达,这主要取决于甲基化位点和甲基化水平(添加的甲基团数)。例如,H3K9me2/3、H3K27me2/3和H4K20me参与基因抑制,而H3K27me1、H3K4me2/3、H3K36me3和H3K79me2/3与基因激活相关联。
5. 组蛋白甲基化与DNA甲基化的相互作用
DNA甲基化是将甲基基团添加到DNA的胞嘧啶碱基上,主要发生在CpG二核苷酸上。DNA甲基化模式与基因沉默和稳定的转录抑制相关联。
组蛋白甲基化与DNA甲基化之间存在广泛的关联和相互作用。某些基因组位点的DNA甲基化依赖于组蛋白甲基化。DNA的修饰状态和序列可以影响染色质中组蛋白蛋白质的甲基化状态。
Kondo等人证实了H3K9甲基化在基因沉默机制中与DNA甲基化具有协同作用,而H3K4甲基化对抗DNA甲基化引起的基因沉默 [6]。
David Allis等人揭示了DNMT3A优先选择H3K36二甲基化区域以促进基因间DNA甲基化,并发现Sotos综合症患者的血样和NSD1突变肿瘤中发生了基因间DNA甲基化的选择性丧失 [7]。这一发现扩展了H3K36甲基化和DNA甲基化修饰之间精确调控机制的认识,对于理解相关人类遗传疾病的发生具有重要的生物学意义。
6. 组蛋白甲基化与疾病
不同位点和状态的组蛋白甲基化可以演变为多种甲基化修饰模式,为组蛋白甲基化调控的基因表达增加了复杂性和多样性。组蛋白甲基转移酶(HMTs)和组蛋白去甲基酶(HDMs)精细地维持了组蛋白甲基化和去甲基化之间的平衡。包括突变或改变组蛋白甲基修饰酶和甲基结合蛋白表达在内的任何改变,都可能扰乱这种平衡,导致各种疾病,特别是癌症 [8]。
例如,H3K27me3甲基转移酶在多种癌症中上调,包括前列腺癌、乳腺癌和淋巴瘤 [9-11]。组蛋白甲基转移酶NSD1和EZH2在许多肿瘤中过度表达,而DOT1L在白血病中发挥重要作用。组蛋白去甲基酶KDM1A和KDM5B分别在恶性程度较高的神经母细胞瘤和前列腺癌中过度表达。LSD1与p53的直接相互作用降低了p53的活性,导致p21表达减少,从而诱导肿瘤发生。甲基化阅读蛋白的异常活动也与许多人类疾病相关,包括发育障碍和癌症。
组蛋白甲基化参与了从受精前到出生后的发育基因表达。甲基化缺陷影响各种发育过程,可以导致成熟动物的发育停滞和致死,或者根据甲基化缺陷的性质和细胞类型特异性导致器官功能的特定缺陷。例如,KDM6B缺失导致心肌细胞增殖在后期发育时停止。完全删除H3K4me3去甲基酶KDM5C导致小鼠中的神经发育异常,并且阻碍了皮质发育。
参考文献:
[1] Strahl BD, Ohba R, Cook RG, Allis CD. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena [J]. Proc Natl Acad Sci U S A. 1999;96:14967–14972.
[2] Rea S, Eisenhaber F, O’Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases [J]. Nature. 2000;406:593–599.
[3] Klose, R. J., and Zhang, Y. (2007). Regulation of histone methylation by demethylimination and demethylation [J]. Nat. Rev. Mol. Cell Biol. 8, 307–318.
[4] Martin C, Zhang Y. The diverse functions of histone lysine methylation [J]. Nat Rev Mol Cell Biol 2005;6:838–849.
[5] E.L. Greer, Y. Shi. Histone methylation: A dynamic mark in health, disease and inheritance [J]. Nat. Rev. Genet., 13 (2012), pp. 343-357.
[6] Kondo Y, Shen L. Issa J P. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer [J]. Mol Cell Biol. 2003. 23(1); 206~215.
[7] David Allis et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape [J]. Nature. 573: 281-286 (2019).
[8] Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers [J]. Nature reviews. Cancer. 2010;10:457–69.
[9] Varambally S, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer [J]. Nature. 2002;419:624–9.
[10] Kleer CG, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells [J]. Proc Natl Acad Sci U S A. 2003;100:11606–11.
[11] Visser HP, et al. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma [J]. British journal of haematology. 2001;112:950–8.





