组蛋白修饰检测
组蛋白后转录修饰(PTMs),也简称为组蛋白修饰,包括甲基化、乙酰化、磷酸化等,对于染色体包装、转录调控以及DNA损伤和修复等各种生物活动起着重要作用 [1-4]。因此,准确敏感地检测组蛋白修饰对于理解生物过程的表观遗传调控和组蛋白修饰酶(HME)靶向药物的开发具有重要意义。
组蛋白修饰检测在揭示表观遗传调控的关键机制中起着重要作用。通过了解组蛋白修饰的重要性、原理和检测方法,我们可以更好地理解组蛋白修饰在基因调控、发育和疾病中的关键机制。
1. 组蛋白修饰检测方法
组蛋白修饰分析是一系列实验方法,用于分析和确定组蛋白的修饰状态和位置,主要包括染色质免疫沉淀(CHIP)和质谱(MS)。这些方法利用特定的实验策略和工具,检测带有特定修饰的组蛋白分子,并定量确定它们的存在量和分布。
1.1 染色质免疫沉淀实验(ChIP)
ChIP是一种常用的组蛋白修饰检测技术,用于检测修饰的存在及其与基因表达的关联。该技术使用特异性抗体结合修饰的组蛋白,将修饰的组蛋白从细胞或组织中富集出来。然后,可以通过PCR、测序或质谱等方法对富集的修饰组蛋白进行进一步分析,确定修饰的存在和定位。
在检测组蛋白修饰方面,有三种主要的基于ChIP的方法,包括ChIP-chip、ChIP-SAGE和ChIP-Seq。
在ChIP中,有两种类型的ChIP:天然ChIP和交联ChIP。天然ChIP使用通过核酸酶消化细胞核制备的天然染色质。交联ChIP使用甲醛固定的染色质,并通过声波破碎进行断裂。在表1中,我们对比了这两种ChIP方法。
表1: 天然ChIP和交联ChIP的比较
| 类型 | 类型 | 劣势 |
|---|---|---|
| 天然ChIP |
|
|
| 交联ChIP |
|
|
此外,您可以点击以下链接查看ChIP操作具体流程:
1.1.1 ChIP-chip
ChIP-chip(染色质免疫沉淀结合DNA微阵列杂交)是一种将染色质免疫沉淀与DNA微阵列相结合的技术。与常规的ChIP一样,ChIP-chip用于研究体内蛋白质与DNA的相互作用。实际上,ChIP-chip方法可用于研究许多表观基因组现象。这里出现的示例展示了ChIP-chip在研究组蛋白修饰中的应用。正如图1所示:
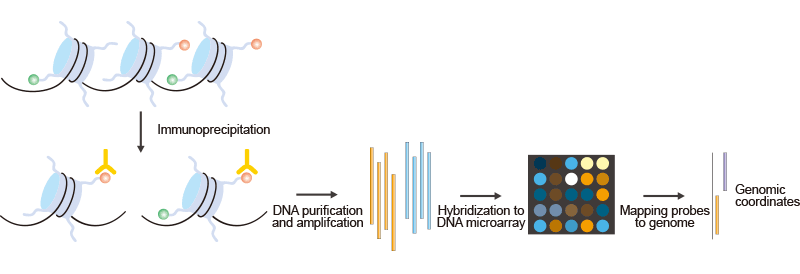
图1. ChIP-on-chip的简要步骤
首先,通过使用特定于特定组蛋白修饰的抗体免疫沉淀交联染色质来纯化修饰的染色质。然后,DNA被扩增以获得足够的DNA(用一种颜色标记)。用颜色标记的ChIP DNA与从输入染色质中准备的控制DNA混合,并用不同颜色标记。随后,微阵列探针可以映射到基因组上,得出基因组坐标。然而,大多数ChIP-on-chip实验仍然使用多克隆抗体,这些抗体在批次之间的特异性不同。因此,生成和使用单克隆抗体进行ChIP-on-chip实验是一个重要目标。
1.1.2 ChIP-SAGE
ChIP-SAGE是指染色质免疫沉淀结合串联分析基因表达。将ChIP实验与SAGE结合,可以在基因组范围内描述组蛋白修饰。ChIP-SAGE过程始于ChIP步骤,用于纯化与特定组蛋白修饰相关的染色质区域,然后按照以下步骤进行。如图2所示:

图2. ChIP-SAGE的简要步骤
首先,交联被逆转,接着将生物素化的通用连接器(UL)连接到DNA末端,并将DNA结合到链霉亲和素珠上。然后,使用识别CATG的Nla III酶消化DNA,将包含Mme I识别序列的连接器连接到切割的DNA末端。Mme I酶消化产生来自免疫沉淀片段的21-22 bp序列标签;这些序列标签被连接在一起,克隆到测序载体中,然后进行测序。每个测序反应中通常可以生成大约20到30个21 bp的短序列标签。然后,这些序列标签可以映射到基因组上,以识别修饰区域。
1.1.3 ChIP-Seq
ChIP-seq(染色质免疫沉淀结合高通量测序技术)是近年来在基因组尺度上研究表观遗传现象的最令人兴奋的技术之一,它依赖于将ChIP实验与高通量测序技术相结合。ChIP-seq将ChIP与高通量DNA测序相结合,用于鉴定DNA相关蛋白的结合位点。如图3所示:

图3. ChIP-Seq的简要步骤
这里概述的过程是针对使用Illumina Genome Analyzer和Solexa技术的。它可以分为两个部分,ChIP和测序。第一步是使用特定于特定组蛋白修饰的抗体对修饰的染色质进行免疫沉淀。然后,修饰的ChIP DNA末端经修复,并与一对适配器连接,随后进行有限的PCR扩增。DNA分子结合到一个包含共价结合的寡核苷酸的流动细胞表面,这些核苷酸可以识别适配器序列。经过尺寸选择,所有产生的ChIP-DNA片段都可以使用基因组测序仪同时进行测序。得到的序列读数被映射到参考基因组上,以获得与免疫沉淀片段对应的基因组坐标。
1.2 质谱法(MS)
质谱法近年来已成为鉴定和定量组蛋白修饰的首选方法。通过质谱法鉴定和定位特定残基的组蛋白修饰依赖于肽或蛋白质的实验测量质量和预期质量之间的"质量差"。理论上,这种方法使得在单次分析中可以对任何PTM或PTM组合进行概要分析,而不受PTM类型或其特定位点的限制,并提供高度准确的定量数据。
质谱法已被证明是评估和比较不同样本中特定修饰及其组合水平的有价值工具。
在生物样本中,有三种基于质谱法的方法可用于检测组蛋白修饰,包括“自上而下”、“中间范围”和“自下而上”的质谱法 [5-8]。
1.2.1 “自上而下”方法
在“自上而下”方法中,完整的组蛋白被色谱分离,然后被离子化并进行质谱分析 [5,8]。由于完整组蛋白的高电荷状态,可以从13+到25+不等,所以高分辨率的质谱和质谱/质谱对于这种技术至关重要。这个过程提供了在给定样本中所有组蛋白同工型以及它们的总体化学计量学的详细数据。
1.2.2 “中间范围”方法
在“中间范围”方法中,通过Glu-C或Asp-N的消化生成长的组蛋白肽,通常分子量超过5 kDa(>20个氨基酸)[5,8]。通过这种方法,可以获得完整的N-末端尾巴。例如,Asp-N产生H4 1–24肽,而Glu-C产生H3 1–50肽,其中包括了H4和H3中大多数著名PTM位点。尽管这两种方法适合于检测长程PTM关联并估计其丰度,但是这两种方法面临的一个主要困难是区分异构肽(具有相同修饰但位于不同位置的肽段),此外,这些方法通常面临灵敏度降低和计算密集型数据分析的困扰。因此,“自上而下”和“中间范围”质谱法通常只在少数专业实验室中进行,并且截至目前尚未有在临床样本中应用的记录。
1.2.3 “自下而上”方法
在“自下而上”质谱法中,组蛋白在通过质谱分析之前,经过Arg‐C蛋白酶介导的酶促消化为相对较短的肽(长度为5-20个氨基酸)。由于核心组蛋白富含碱性氨基酸残基,胰蛋白酶产生的肽太短,并且在修饰位点附近低效切割,导致长度不一致,不适合准确定量。在胰蛋白酶切割之前,通过氘化乙酸酐或丙酸酐在N-末端和赖氨酸上的游离胺衍生通常用于模拟Arg-C消化,同时使用强大的胰蛋白酶 [9-12]。这种策略也有助于区分等压肽 [10]。
虽然“自下而上”提供有限的同时发生的PTM数据(最多四个),特别是对于远处的标记,但它提供了灵活性,并在分析患者来源的样本中具有应用。
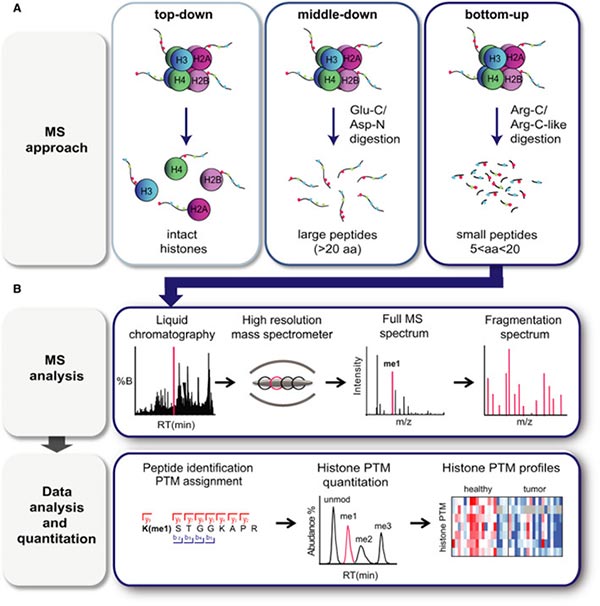
图4. 基于质谱法的组蛋白修饰分析
图片引用自: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9291046/
2. 组蛋白修饰检测的应用
组蛋白修饰检测在表观遗传学研究和个体化医学中至关重要。它有助于理解基因调控机制,探索细胞分化和组织发育过程,识别疾病生物标志物,并开发靶向治疗方法。
2.1 揭示基因调控机制
组蛋白修饰检测可以帮助我们揭示基因调控机制。通过分析基因启动子和增强子中不同修饰的分布,我们可以了解组蛋白修饰在基因表达中的调节作用,并进一步了解基因调控的详细机制。
2.2 研究发育和细胞命运决定
组蛋白修饰检测在发育过程和细胞命运决定中发挥重要作用。通过分析修饰的动态变化,我们可以研究细胞分化和组织发育的分子机制,以及干细胞命运的调控。
2.3 探索疾病发病机制
组蛋白修饰的检测帮助我们了解疾病的机制。不同疾病中异常的组蛋白修饰模式可以为疾病的发生和发展提供重要线索,从而为疾病的诊断和治疗提供新的靶标和策略。
参考文献:
[1] Valensisi, C., Liao, J.L., Andrus, C. et al. cChIP-seq: a robust small-scale method for investigation of histone modifications [J]. BMC Genomics 16, 1083 (2015).
[2] Peterson CL, Laniel MA. Histones and histone modifications [J]. Curr Biol. 2004;14(14):R546–51.
[3] Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications [J]. Cell Res. 2011;21(3):381–95.
[4] Binder H, Steiner L, et al. Transcriptional regulation by histone modifications: towards a theory of chromatin re-organization during stem cell differentiation [J]. Phys Biol. 2013;10(2):026006.
[5] O’Geen H, Echipare L, Farnham PJ. Using ChIP-Seq technology to generate high-resolution profiles of histone modifications [J]. Methods Mol Biol. 2011;791:265–86.
[6] Noberini R, Robusti G, Bonaldi T. Mass spectrometry-based characterization of histones in clinical samples: applications, progress, and challenges [J]. FEBS J. 2022 Mar;289(5):1191-1213.
[7] S. Sidoli, B.A. Garcia. Characterization of individual histone posttranslational modifications and their combinatorial patterns by mass spectrometry-based proteomics strategies [J]. Methods Mol. Biol., 1528 (2017), pp. 121-148.
[8] Z.-F. Yuan, A.M. Arnaudo, B.A. Garcia. Mass spectrometric analysis of histone proteoforms [J]. Annu. Rev. Anal. Chem., 7 (2014), pp. 113-128.
[9] A. Moradian, A. Kalli, M.J. Sweredoski, S. Hess. The top-down, middle-down, and bottom-up mass spectrometry approaches for characterization of histone variants and their post-translational modifications [J]. Proteomics, 14 (2014), pp. 489-497.
[10] Smith CM, Haimberger ZW, et al. (2002) Heritable chromatin structure: mapping "memory" in histones H3 and H4 [J]. Proc Natl Acad Sci USA 99 (Suppl 4), 16454–16461.
[11] Soldi M, Cuomo A & Bonaldi T (2014) Improved bottom‐up strategy to efficiently separate hypermodified histone peptides through ultra‐HPLC separation on a bench top Orbitrap instrument [J]. Proteomics 14, 2212–2225.
[12] Sidoli S, Bhanu NV, Karch KR, Wang X & Garcia BA (2016) Complete workflow for analysis of histone post‐translational modifications using bottom‐up mass spectrometry: from histone extraction to data analysis [J]. J Vis Exp 111, 54112.





